Votre maladie
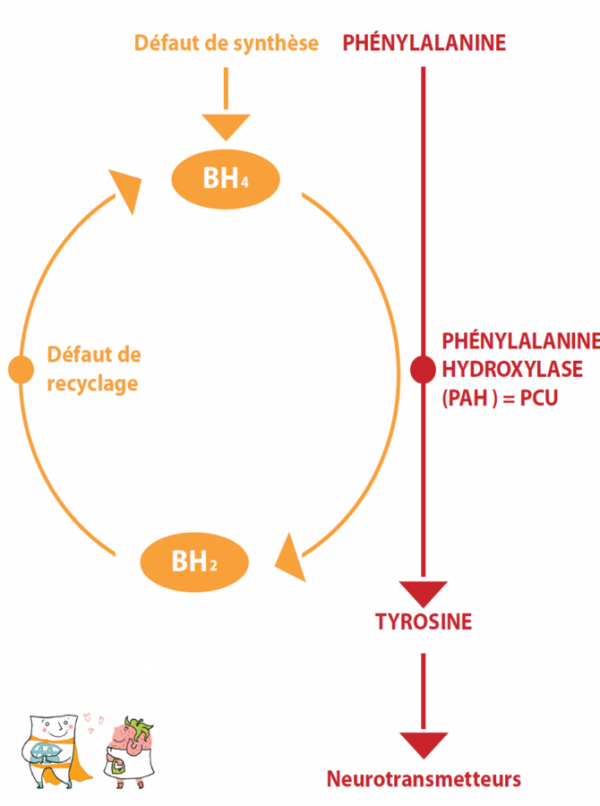
La Phénylcétonurie (PCU) est due au déficit d’une enzyme : la phénylalanine hydroxylase (PAH) permettant de transformer la phénylalanine en Tyrosine. C’est la 1ère maladie métabolique héréditaire traitée par un régime. Elle est donc bien connue et le traitement diététique instauré précocement a fait ses preuves. C’est pourquoi un dépistage néonatal systématique existe en France depuis 1975. La fréquence de la PCU est d’environ 1/16 000 naissances en France.
Petit rappel biochimique et conséquences du déficit enzymatique
Selon l’activité enzymatique résiduelle de la phénylalanine hydroxylase, la tolérance en phénylalanine varie :
- lorsque les taux sanguins de phénylalanine, avec une alimentation normale demeurent supérieurs à 20mg/100ml (1200 μmol/l), la PCU est appelée PCU classique (ou typique) avec une activité enzymatique nulle et une tolérance habituellement de l’ordre de 250 à 300mg de phénylalanine/jour.
- Lorsque les taux sanguins sont entre 10 et 20mg/100ml (600 à 1200 μmol/l), la PCU est appelée atypique, avec une activité enzymatique résiduelle faible, mais permettant une augmentation de la tolérance (jusqu’à 600 à 1000mg de phénylalanine/jour).
- Lorsque les taux sanguins sont inférieurs à 10mg/100ml (600 μmol/l), on parle d’ Hyperphénylalaninémie modérée permanente et il n’y a pas lieu habituellement de faire un régime. Il faudra avoir des apports normoprotidiques.
Cette classification a toutefois ses limites, car elle est basée sur le taux sanguin de phénylalanine en régime normal, c’est-à-dire riche en phénylalanine.
Ainsi, un enfant en allaitement maternel exclusif peut être considéré initialement comme ayant une hyperphénylalaninémie modérée (taux <10 mg/100ml, le lait maternel étant plus pauvre en phénylalanine que le lait artificiel), alors que sa concentration sanguine de phénylalanine augmentera au-delà de 10 mg/100ml lors du sevrage de l’allaitement maternel, le faisant finalement classer comme une PCU atypique.
De plus, il peut exister à la naissance un certain degré d’immaturité de l’enzymeUne enzyme est une protéine (dans certains cas, un ARN) qui va catalyser (accélérer) une réaction du métabolisme. phénylalanine hydroxylase, et une amélioration de la tolérance en phénylalanine reste possible les premières années de vie.
Il peut exister d’autres formes de PCU.
Dans les conditions normales,cette enzymeUne enzyme est une protéine (dans certains cas, un ARN) qui va catalyser (accélérer) une réaction du métabolisme. a besoin d’un cofacteurComme son nom l’indique, un cofacteur intervient avec d’autres molécules (protéines, vitamines…) au cours d’une réaction. Sa présence peut être indispensable au bon déroulement du processus. : la BH4 (Tétrahydrobioptérine), pour fonctionner correctement. Il peut aussi exister des formes de PCU sensibles à la BH4. Dans ces rares cas, l’ajout de BH4 par voie orale sous forme de médicament permet d’augmenter l’activité enzymatique résiduelle, avec comme conséquences la possibilité d’élargir franchement le régime (amélioration de la tolérance en phénylalanine), voire d’arrêter complètement la prise du mélange d’acides aminés.
La sensibilité à la BH4 concerne principalement les PCU atypiques, et reste exceptionnelle pour les PCU classiques. Il faut donc « tester cette sensibilité » pour s’assurer d’une réponse positive et d’un traitement médicamenteux potentiel. Dans le cas d’une phénylcétonurie sensible et traitable par la BH4, le traitement sera alors « médicamenteux » ; la forme commerciale de la BH4 existe depuis 2009 en France, elle est appelée KUVAN et est remboursée à 100% par la Sécurité Sociale, avec une autorisation de mise sur le marché (AMM) pour les enfants de plus de 4 ans et déconseillé pendant les grossesses. Les discussions actuelles portent sur l’intérêt réel d’un traitement par la BH4 lorsqu’il ne permet qu’un élargissement modeste du régime.
Enfin, il existe d’authentiques déficits de synthèse de la BH4, qui se révèlent par une hyperphénylalaninémie au moment du dépistage néonatal. Dans ce cas, il ne s’agit pas à proprement parler d’une « vraie phénylcétonurie » par déficit en phénylalanine hydroxylase, mais d’un déficit de son cofacteurComme son nom l’indique, un cofacteur intervient avec d’autres molécules (protéines, vitamines…) au cours d’une réaction. Sa présence peut être indispensable au bon déroulement du processus., la BH4, qui intervient également comme cofacteurComme son nom l’indique, un cofacteur intervient avec d’autres molécules (protéines, vitamines…) au cours d’une réaction. Sa présence peut être indispensable au bon déroulement du processus. d’autres réactions enzymatiques. Le traitement et la surveillance ultérieure sont alors différents et ne seront pas abordés ici.
Envisageons le cas d’un enfant sous régime hypoprotidique, dont le principe correspond à celui indiqué dans les principes généraux mais qui vous sera expliqué en détails par l’équipe de votre centre de soins, et abordons diverses questions qui pourront se présenter à vous lors de votre retour à la maison.