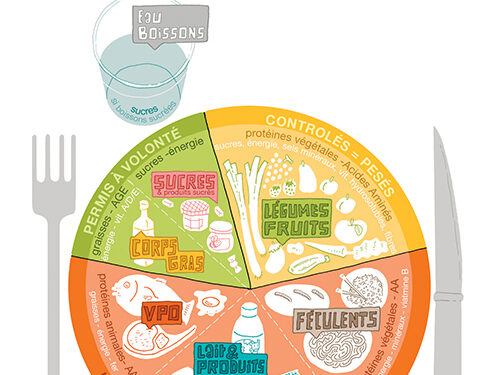
La phénylcétonurie
La phénylcétonurie est une maladie génétique appartenant aux maladies nommées « les erreurs innées du métabolisme des acides aminés », ce sont des maladies orphelines.
La phénylcétonurie a été décrite pour la première fois en 1934 par le Docteur Norvégien AsbjØrn FØlling, mais ce n’est que 20 ans plus tard, en 1954, que le Professeur Horst Bickel évoque la mise en place d’un régime pauvre en phénylalanine. Il faudra attendre 1962, pour que le Docteur Robert Guthrie, mette au point le dépistage.