Your disease
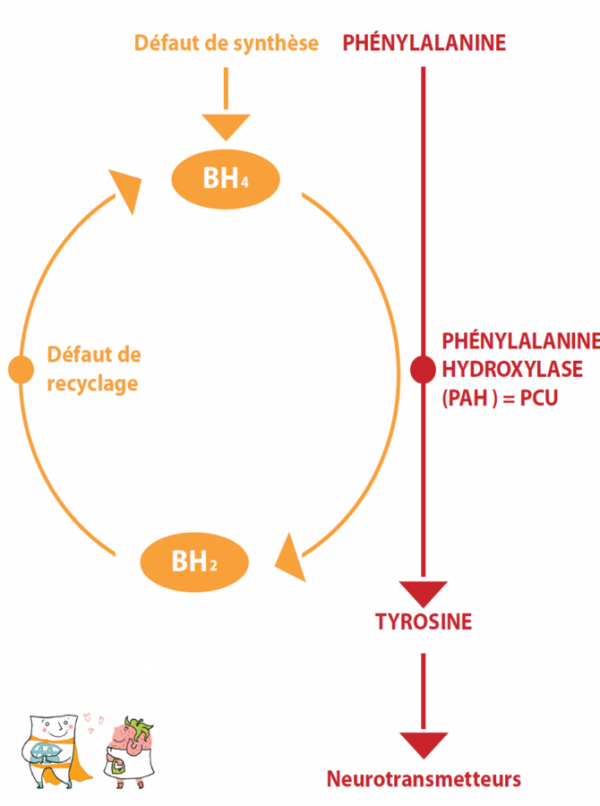
Phenylketonuria (PKU) is due to the deficiency of an enzyme: phenylalanine hydroxylase (PAH), which converts phenylalanine into tyrosine. It is the first hereditary metabolic disease to be treated by diet. It is therefore well known and early dietary treatment has proved successful. This is why systematic neonatal screening has existed in France since 1975. The frequency of PKU is approximately 1/16,000 births in France.
Brief biochemical reminder and consequences of the enzyme deficiency
Depending on the residual enzymatic activity of phenylalanine hydroxylase, phenylalanine tolerance varies:
- When blood levels of phenylalanine, with a normal diet, remain above 20mg/100ml (1200 μmol/l), PKU is termed classical (or typical) PKU with zero enzymeAn enzyme is a protein (or in certain cases an RNA) which catalyses (speeds up) a metabolic reaction. activity and a tolerance usually in the range of 250-300mg phenylalanine/day.
- When blood levels are between 10 and 20mg/100ml (600 to 1200 μmol/l), ECP is termed atypical, with low residual enzymeAn enzyme is a protein (or in certain cases an RNA) which catalyses (speeds up) a metabolic reaction. activity, but allowing for increased tolerance (up to 600 to 1000mg phenylalanine/day).
- When blood levels are below 10mg/100ml (600 μmol/l), this is termed permanent moderate hyperphenylalaninaemia and there is usually no need for diet. Normoprotein intakes should be maintained.
This classification has its limitations, however, as it is based on the blood level of phenylalanine in the normal, i.e. phenylalanine-rich diet.
Thus, an exclusively breastfed child may initially be considered to have moderate hyperphenylalaninaemia (<10 mg/100ml, as breastmilk is lower in phenylalanine than artificial milk), whereas his or her blood phenylalanine concentration will increase beyond 10 mg/100ml upon weaning from breastmilk, ultimately classifying him or her as atypical PKU.
In addition, there may be some degree of immaturity of the enzymeAn enzyme is a protein (or in certain cases an RNA) which catalyses (speeds up) a metabolic reaction. phenylalanine hydroxylase at birth, and improvement in phenylalanine tolerance is still possible in the first years of life.
There may be other forms of ECP.
Under normal conditions, this enzymeAn enzyme is a protein (or in certain cases an RNA) which catalyses (speeds up) a metabolic reaction. needs a cofactorAs its name indicates, a cofactor intervenes with other molecules (proteins, vitamins, etc.) during a reaction. Its presence may be essential for the process to run smoothly. (see general principles), BH4 (Tetrahydrobiopterin), to function properly. There may also be forms of ECP that are sensitive to BH4. In these rare cases, the addition of oral BH4 in the form of a drug increases the residual enzymatic activity, with the result that the diet can be significantly extended (improvement of phenylalanine tolerance), or even the intake of the amino acidAmino acids are molecules that combine to form proteins. 20 amino acids make up the proteins of the human body. Of these, 8 are essential (our body cannot synthesise them, they must be supplied by the diet): isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine.
Arginine and histidine are semi-indispensable. In fact, only infants need to take them from their food.
Cysteine, glycine and tyrosine may be indispensable for certain populations mixture can be stopped completely.
Sensitivity to BH4 occurs mainly in atypical PKU, and is exceptional in classical PKU. It is therefore necessary to “test for sensitivity” to ensure a positive response and potential drug treatment. In the case of sensitive phenylketonuria treatable by BH4, the treatment will be “medicated”; the commercial form of BH4 has existed since 2009 in France, it is called KUVAN and is reimbursed at 100% by the French Social Security, with a marketing authorisation (AMM) for children over 4 years of age and not recommended during pregnancy. The current discussion concerns the real interest of a treatment with BH4 when it only allows a modest extension of the diet.
Finally, there are genuine deficits in the synthesis of BH4, which are revealed by hyperphenylalaninemia at the time of neonatal screening. In this case, it is not strictly speaking a “true phenylketonuria” due to phenylalanine hydroxylase deficiency, but a deficiency of its cofactorAs its name indicates, a cofactor intervenes with other molecules (proteins, vitamins, etc.) during a reaction. Its presence may be essential for the process to run smoothly., BH4, which also acts as a cofactorAs its name indicates, a cofactor intervenes with other molecules (proteins, vitamins, etc.) during a reaction. Its presence may be essential for the process to run smoothly. in other enzymatic reactions. The treatment and subsequent monitoring is therefore different and will not be discussed here.
Let’s consider the case of a child on a hypoprotein diet, which corresponds to the general principles but which will be explained to you in detail by your health centre team, and let’s address various issues that may arise when you go home.